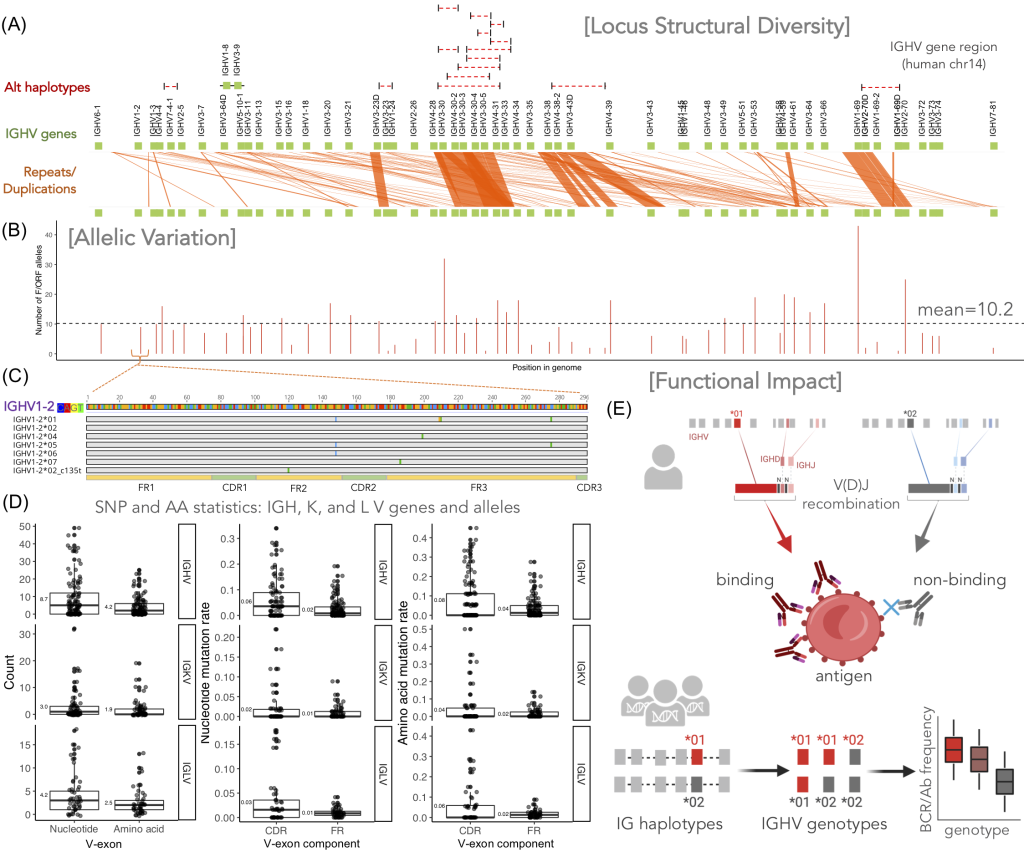
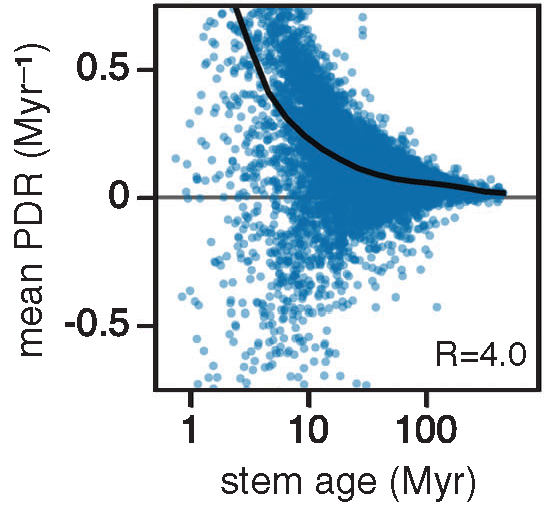
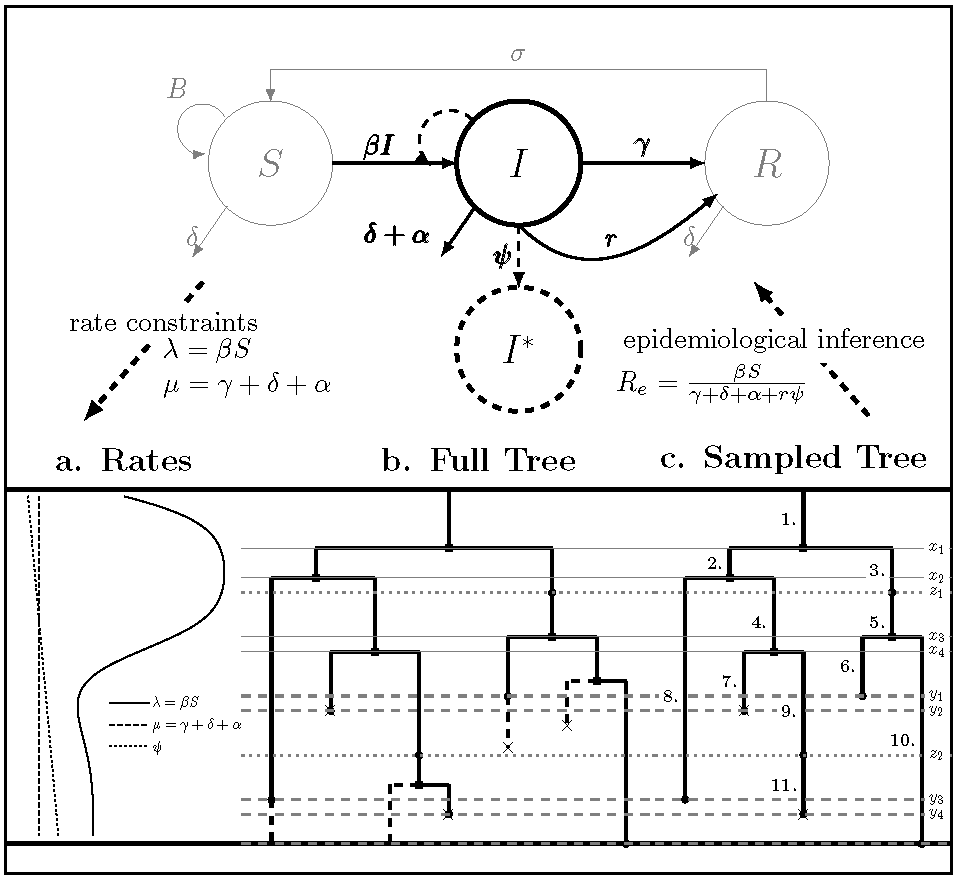
Our team uses phylogenetic trees, graphical depictions of historical relationships, to study evolution at multiple scales—from the grandest (e.g., the origin of major groups of organisms) to the smallest (e.g., the development of the adaptive immune system within an individual) of scales. In our research, we develop computational and statistical tools, analyze mathematical models, and conduct large-scale empirical analyses to answer evolutionary questions.